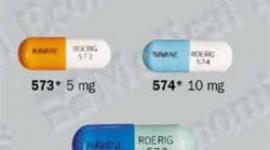
Fetzima (levomilnacipran) Patient Information
Brand Name: Fetzima
Generic Name: Levomilnacipran
Fetzima (levomilnacipran) Full Prescribing Information
Fetzima Medication Guide
Information for Patients
Advise patients, their families, and their caregivers about the benefits and risks associated with treatment with Fetzima and counsel them on its appropriate use. Advise patients, their families, and their caregivers to read the Medication Guide and assist them in understanding its contents.
Suicide Risk
Advise patients and caregivers to look for the emergence of suicidality, especially early during treatment and when the dose is adjusted up or down.
Dosing and Administration
Advise patients that Fetzima should be swallowed whole and should not be chewed, crushed or opened.
Advise patients that Fetzima can be taken with or without food. Fetzima should be initiated with a dose of 20 mg once daily for 2 days and then increased to 40 mg once daily. Based on efficacy and tolerability, Fetzima may then be increased in increments of 40 mg at intervals of 2 or more days. The maximum recommended dose is 120 mg once daily.
Instruct patients if they miss a dose, to take the missed dose as soon as they remember. If it is almost time for the next dose, instruct them to skip the missed dose and take their next dose at the regular time. Advise them not to take two doses of Fetzima at the same time.
Concomitant Medication
Instruct patients not to take Fetzima with an MAOI or within 14 days of stopping an MAOI and to allow 7 days after stopping Fetzima before starting an MAOI.
Serotonin Syndrome
Caution patients about the risk of serotonin syndrome, particularly with the concomitant use of Fetzima and triptans, tramadol, tryptophan supplements, other serotonergic agents, or antipsychotic drugs.
Effect on Blood Pressure and Heart Rate
Advise patients that they should have regular monitoring of blood pressure and heart rate when taking Fetzima.
Abnormal Bleeding
Caution patients about the concomitant use of Fetzima and NSAIDs, aspirin, warfarin, or other drugs that affect coagulation since combined use of psychotropic drugs that interfere with serotonin reuptake and these agents has been associated with an increased risk of abnormal bleeding.
Narrow-angle Glaucoma
Advise patients with raised intraocular pressure or those at risk of acute narrow-angle glaucoma (angle-closure glaucoma) that mydriasis has been reported with Fetzima and they should be monitored.
Urinary Hesitation or Retention
Caution patients about the risk of urinary hesitation and retention while taking Fetzima, particularly in patients prone to obstructive urinary disorders.
Activation of Mania/Hypomania
Advise patients and their caregivers to observe for signs of activation of mania/hypomania.
Seizures
Caution patients about using Fetzima if they have a history of a seizure disorder. Patients with a history of seizures were excluded from clinical studies.
Discontinuation Syndrome
Advise patients not to stop taking Fetzima without first talking with their healthcare provider. Patients should be aware that discontinuation effects may occur when suddenly stopping Fetzima.
Hyponatremia
Advise patients that if they are treated with diuretics, or are otherwise volume depleted, or are elderly, they may be at greater risk of developing hyponatremia while taking Fetzima.
Alcohol
Advise patients to avoid consumption of alcohol while taking Fetzima.
Allergic Reactions
Advise patients to notify their healthcare provider if they develop an allergic reaction such as rash, hives, swelling, or difficulty breathing.
Pregnancy
Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during therapy with Fetzima.
Nursing Mothers
Advise patients to notify their healthcare provider if they are breastfeeding an infant and would like to continue or start Fetzima.
Interference with Cognitive and Motor Performance
Caution patients about operating hazardous machinery, including automobiles, until they are reasonably certain that Fetzima therapy does not adversely affect their ability to engage in such activities.
Last revision: 07/2013
Fetzima (levomilnacipran) Full Prescribing Information
Fetzima Medication Guide
APA Reference
Staff, H.
(2013, October 9). Fetzima (levomilnacipran) Patient Information, HealthyPlace. Retrieved
on 2026, July 22 from https://www.healthyplace.com/other-info/psychiatric-medications/fetzima-levomilnacipran-patient-information